To compile our comprehensive dataset of activating mutations in PKs, we performed a two-step systematic search for each of the 518 PKs present in the "complete kinase" study of the COSMIC database. For more information refer to Molina Vila et al, 2014
Kin-Driver database offers a comprehensive set of 560 primary activating mutations (AM) in the kinase and justamembrane (JM) domains of 39 PKs and 83 inactivating mutations in 5 kinases compiled by a two-step systematic search for each of the 518 PKs present in the “complete kinase” study of the COSMIC database (release 70). Detail description of the activating mutations search is found in Molina-Vila et al 2014 (link). Only primary mutations with experimental evidence demonstrating their activating/inactivating role were included. Kin-Driver is a MySQL relational database offering structural and sequence data crossreferenced with COSMIC and with our set of curated mutations. It also provides the frequencies of these mutations in actual tumour samples. The CosmicMart service is used to fetch the data, so frequencies for new mutations can easily be added and data are kept up to date with the periodic COSMIC releases.
The server accepts searches by
The server maps each variant to Uniprotprotein sequences. If the reference residue in the Uniprot protein sequence is different from the one indicated in your variant the analysis will not be performed.
For each variant the server provides the following annotations:
| Column name | Description |
| Uniprot ID | Uniprot protein accession ID |
| Mutation | Amino-acid substitution |
| Relative frequency | Relative frequency of the mutation, calculated by dividing the number of tumors carrying the mutation by the total number of tumors where the corresponding gene has been sequenced (according to COSMIC), and then multiplying the resulting figure by 1000. |
| Validation status | A, Activating; I, Inactivating; UNK, unknown effects |
| Reference | PubMed reference demonstrating by experimental data that the mutation is activating/inactivating. No annotation will appear here if the effects of the mutation are unknown |
| Type | Missense (single amino-acid substitution), nonsense (point mutation generating a stop codon), indel (small insertions, deletions or insertions/deletions) |
| MSA | Link to Multiple Sequence Alignment (MSA) browser. MSA will only be available for mutations in the kinase domain. |
| PDB | Link to 3D structure. |
Our server allows to determine if a mutation falls within one of the three "hypermutated segments" clustering activating mutations in PKs.
Activating mutations of kinases do not associate with conserved positions and do not usually affect ATP binding or catalytic residues. Instead, they cluster around three "molecular brakes" of the kinase activity, namely the αC helix plus the hinge, the activation loop, and in the PDGFR subfamily, the juxtamembrane region.
In Tyrosin Kinases (TKs), 225 activating mutations in 22 proteins are located in the kinase domain. We built a multiple sequence alignment (MSA) of TKs and then mapped these mutations onto a MSA and projected the MSA of the entire TK family onto the 3D structure of the human EGFR. We selected EGFR as a reference because it is a well-established genetic driver of tumorigenesis in several human tumor types, is a TK in which numerous activating mutations have been described, and has been characterized structurally in numerous crystallographic studies. An accumulated frequency of activating mutations was then calculated for each position in the aminoacidic sequence and subsequently normalized. We found that 154 of the 225 mutations (68%) were clustered in two hypermutated segments (HS2 and HS3). This clustering was apparent both in terms of normalized frequencies and in terms of the absolute number of different mutations and TKs affected. The distribution could also be visualized in a central moving average plot, which gave a two-peak profile. In contrast, when we mapped the hundreds of COSMIC mutations in the TKs under study that have not been described as activating, they were almost uniformly scattered within the kinase domain. The distribution of these putative passenger mutations was significantly different from that of the activating mutations by both the Wilcoxon and the Kolmogorov–Smirnov tests (p<2.2 10-16), and the clustering of the activating mutations in the HS2 and HS3 was statistically significant in a two-tailed Fisher’s exact text (p<0.0001) when compared to the putative passenger mutations.
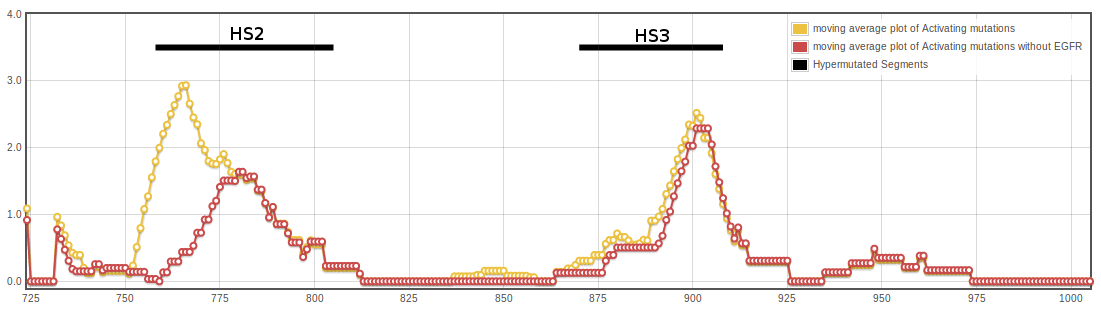
The HS2, which clusters 86 activating mutations in 16 different kinases, comprises the C-terminal half of the subdomain SDII, the entire SDIII (αC-helix) plus SDIV (kinase hinge), and the N-terminal half of the SDV. The HS3, which clusters 68 mutations in 14 TKs, includes the subdomains VIB (catalytic loop), VII (activation loop) and the two N-terminal residues of VIII (P+I loop). In the HS3 segment, a hotspot in position 861 clusters 21 activating mutations in six different TKs. In silico studies have shown that both HS2 and HS3 are regions where nonsynonymous, cancer-associated single-base changes in PKs are preferentially located. The EGFR TK alone accounts for 71 mutations in the kinase domain. When we compared the distribution of activating mutations within the HS2 including and excluding EGFR, the Spearman’s coefficient showed a weak correlation (0.3). While 30 activating mutations are present in the SDII of EGFR, only one is present in the remaining 20 TKs. Thus, activating mutations in the SDII, most of which are in-frame insertions/deletions, seem to be a particularity of EGFR. In addition, EGFR accounts for half of the activating mutations in the catalytic loop (VIB region). In consequence, if EGFR is excluded, the two HS could be redefined to comprise fewer amino acids
One hundred and twenty-one activating mutations were located in the JM region of the four RTKs analyzed belonging to the PDGFR family, plus EGFR and RET, 72 of which corresponded to KIT and 34 to FLT3. Twelve additional mutations mapped to the first nine residues of the kinase domain. This led us to define another hypermutated segment (HS1), comprising the JM region plus the N-terminus of the SDI (Tables 1 and 2). The JM region of the four PDGFR family receptors was aligned, and activating mutation positions were mapped onto the KIT structure. Most of the mutations were located in residues 551–578. EGFR and RET were excluded from the alignment due to the significant differences in the JM amino acid sequence.
In STKs, 97 mutations in 13 different proteins were located within the kinase domain. In this case, we used human BRAF as a reference structure in the MSA. Due to the low number of activating mutations reported in STKs, statistical analysis was impossible and caution must be exercised when analyzing our results. However, activating mutations in STKs also seem to cluster in the HS3 segment defined for TKs (10 mutations) and, to a lesser extent, in HS1 and HS2 (five and four mutations, respectively) . In addition, the most frequently mutated residue in BRAF (V600) overlaps with the 861 activating mutation hotspot of TKs.